
 Wunde, Wundheilung
Wunde, Wundheilung Infektion
Infektion Akutes Abdomen
Akutes Abdomen Abdominaltrauma
Abdominaltrauma Ileus
Ileus Hernien
Hernien- Allgemeine Chirurgie
 Struma benigna
Struma benigna Schilddrüsen-CA
Schilddrüsen-CA Nebenschilddrüsen
Nebenschilddrüsen Hypderthyreose
Hypderthyreose Nebenniere
Nebenniere- Endokrine Chirurgie
 Achalasie
Achalasie Ösophagus-CA
Ösophagus-CA Ösophagusdivertikel
Ösophagusdivertikel Ösophagusperforation
Ösophagusperforation Verätzung
Verätzung Magen-CA
Magen-CA Ulkuskrankheit
Ulkuskrankheit GERD
GERD Adipositas
Adipositas- Oberer GI-Trakt
 CED
CED Divertikulitis
Divertikulitis Kolon-CA
Kolon-CA Proktologie
Proktologie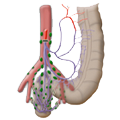 Rektum-CA
Rektum-CA- Unterer GI-Trakt
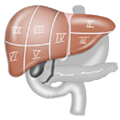 Aantomie
Aantomie Ikterus
Ikterus Cholezystolithiais
Cholezystolithiais Benigne Leberläsionen
Benigne Leberläsionen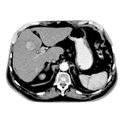 Maligne Leberläsionen
Maligne Leberläsionen Pankreatitis
Pankreatitis Pankreaskarzinom
Pankreaskarzinom- Hepatobiliäre Chirurgie
Infektion
Eine Infektion ist definiert als das Eindringen von Mikroorganismen in den Körpern bei gleichzeitig vorliegender lokalisierter oder generalisierter Reaktion des Körpers.
Die Erreger lassen sich dabei in obligat pathogen und fakultativ pathogen einteilen. Obligat pathogene Keime verursachen dabei in jedem Fall Krankheitssymptome, sie gehören nicht zur normalen Körperflora. Als Beispiel ist Samonella tyhphi zu nennen. Fakultativ pathogen wirkt z.B. Eschericha coli. Dieses Bakterium gehört zur normalen Darmflora und verursacht dort keine Infektion. Erst wenn es diesen Besiedelungsort verlässt und beispielsweise in eine Wunde gelangt, spricht man von einer Infektion.
Einer anderen Einteilung folgend spricht man von endogenen oder exogenen Erregern sowie von opportunistischen Erregern. Zu letzteren zählt z.B. Pseudomonas aeruginosa. Dieser Keim nutzt bei einer opportunistischen Besiedelung eine vorbestehende Immunschwäche aus, bei normal funktionierendem Immunsystem würde keine Infektion verursacht.






Morphologie
Morphologisch lassen sich Infektionen nach Ihrem Erscheinungsbild einteilen. Unspezifische Morphologien sind z.B. ein Abszess durch Staphylococcus aureus oder eine Phlegmone. Ein Abszess ist definiert als eitrige Gewebeeinschmelzung, die von umgebenden Organstrukturen durch eine Membran abgegrenzt ist. Eine Phlegmone ist eine tiefe Infektion des Gewebes, die sich diffus ausbreitet, also nicht abgegrenzt ist. Auch ein Empyem, also die Eiteransammlung in einer präformierten Körperhöhle ist eine Infektion mit einer unspezifischen Morphologie.
Granulome bei Mykobakterium tuberculosis, Gummen bei Treponema pallidum und Drusen bei einer Infektion mit Actinomyceten sind Infektionen mit spezifischer Morphologie. Dann gibt es auch Erreger, die ein spezielles Endotoxin bilden, die spezifische Reaktionen am Körper hervorrufen. Dazu gehören die Clostridien mit den Untergruppen C. tetani die den Wundstarrkrampf auslösen, C. perfringens als Verursacher des Gasbrands und C. diphteriae.

Opisthotonus bei einem Patient mit Tetanusinfektion - Gemälde von Charles Bell 1809
Der Wundstarrkrampf ist eine Erkrankung, die in unseren Breiten weitestgehend aus dem Bewusstsein verschwunden ist, da die Krankheit durch die hohe Rate an geimpften Menschen nur sehr selten ausbricht. In den Entwicklungsländern der Erde sterben allerdings viele Menschen an Tetanus-Infektionen. Bei der chirurgischen Wundbehandlung ist die Anamnese und ggf. Auffrischung des Tetanus-Impfstatus ein wichtiger Bestandteil. Ausgelöst wird der Wundstarrkrampf durch Clostridium tetani, ein obligat anaerobes Bakterium, das in seiner Sporenform äußerst resistent ist und im Erdreich ubiquitär vorkommt. Die Bakterienform ist in der Lage, verschiedene Exotoxine zu bilden, u.a. Tetanospasmin und Tetanolysin. Dieses gelangt von der Wunde über axonale Transportmechanismen in die Vorderhörner des Rückenmarks, wo es die typischen Symptome des Wundstarrkrampfs auslöst: Trismus (Kiefersperre), Risus sardonicus (Krämpfe der Gesichtsmuskulatur), Spontankrämpfe, Ophistotonus (Überwiegen der Streckmuskulatur des Rückens).
Eine manifeste Tetanusinfektion kann letztlich über Lähmung der Schlundmuskulatur zu Asphyxie und somit zum Tode führen. Unbehandelt verläuft die Krankheit meist tödlich, eine durch Intensivmedizin überstandene Infektion hinterlässt keine Immunität. Therapeutisch ist eine breite Exzision der Wunde und eine offene Wundbehandlung wichtig. Es werden hochdosiert Antibiotika verabreicht, häufig liegen Mischinfektionen vor. Daneben sollte sofort eine aktive Immunisierung mit Tetanus-Adsorbat (Tetanol) und Gabe von Tetanus-Hyperimmunglobulin (Tetagam) erfolgen. Je nach Schwere der Symptome kann eine Sedierung oder Muskelrelaxation mit Beatmungstherapie notwendig werden.
– Hugo Schottmüller
Eine Sepsis liegt dann vor, wenn sich innerhalb des Körpers ein Herd gebildet hat, von dem konstant oder periodisch pathogene Bakterien in den Blutkreislauf gelangen und zwar derart, dass durch diese Invasion subjektive und objektive Krankheitserscheinungen ausgelöst werden.
Sepsis
Der Begriff der Sepsis wurde schon im Zeitalter von Hippokrates erwähnt, aber erst durch den Arzt Hugo Schottmüller im Jahre 1914 näher definiert: „Eine Sepsis liegt dann vor, wenn sich innerhalb des Körpers ein Herd gebildet hat, von dem konstant oder periodisch pathogene Bakterien in den Blutkreislauf gelangen und zwar derart, dass durch diese Invasion subjektive und objektive Krankheitserscheinungen ausgelöst werden.“ [1]
Gastbeitrag
 Dieser Beitrag über die Sepsis wurde freundlicherweise von Dr. Flemming erstellt
Dieser Beitrag über die Sepsis wurde freundlicherweise von Dr. Flemming erstellt
Sepsis – eine der häufigsten Todesursachen
Die Sepsis ist heutzutage eine gefürchtete Komplikation im klinischen Alltag und ist trotz des ständigen medizinischen Fortschritts in Forschung, Diagnostik und Therapie immer noch mit einer sehr hohen Morbidität und Mortalität verbunden. Auf nicht kardiologischen Krankenhausstationen in Deutschland ist die Sepsis die häufigste Todesursache [2-4]. Durch die epidemiologische Entwicklung der Bevölkerung und durch die Erweiterung der Indikationsstellung invasiver Methoden, sowie durch eine Progredienz von Antibiotikaresistenzen und immunsuppressiven Verfahren ist die Inzidenz der Sepsis jährlich steigend [5, 6]. Das Kompetenznetzwerk Sepsis (SepNet) ermittelte für die Sepsis eine Prävalenz von 12,4% sowie für die schwere Sepsis und den septischen Schock eine Prävalenz von 11%. Die Mortalität schwankt dabei je nach Studie und Schweregrad zwischen 20 bis 60% [5-9]. Nach den Atemwegsinfektionen ist ein intraabdomineller Fokus die häufigste Ursache eines schweren septischen Krankheitsbildes. Weiterhin können vor allem katheter- bzw. fremdkörperassoziierte Infektionen, chirurgische Infektionen, invasive Candidosen bei immunsupprimierten Patienten und bakterielle Meningitiden ursächlich sein für die schwere Sepsis.
SIRS? Sepsis? – Definitionen eines Krankheitsbildes
Nach der anfänglichen Sepsis-Definition durch Hugo Schottmüller wird das Krankheitsbild der Sepsis unter Berücksichtigung des heutigen Wissenstands wie folgt definiert: „Die Sepsis wird ausgelöst durch pathogene Mikroorganismen und deren Produkte und führt zu einer endogenen Bildung und Ausschüttung von humoralen und zellulären Mediatoren. In Abgrenzung zu einer Infektion ist sie dadurch gekennzeichnet, dass es dem Wirt nicht gelingt, die inflammatorische Reaktion lokal zu begrenzen.“ [3]
Neben dieser allgemeinen Definition erfolgte im Jahre 1991 im Rahmen der Consensus Conference des American College of Chest Physicians und der Society of Critical Care Medicine eine weiterreichende Einteilung und Definition der Sepsis, da man im Laufe der Zeit erkennen musste, dass nicht nur infektiologische Stimuli sondern auch z.B. Traumata, Verbrennungen, Intoxikationen und Ischämien ein septisches Krankheitsbild auslösen können [10, 11]. Hier wurde zwischen einer systemischen inflammatorischen Reaktion (systemic inflammatory response syndrome, SIRS), einer Sepsis, einer schweren Sepsis und dem septischen Schock unterschieden. Eine systemische inflammatorische Reaktion (systemic inflammatory response syndrome, SIRS) ist gekennzeichnet durch das Vorliegen von mindestens zwei der 4 nachfolgenden Kriterien:
- Körpertemperatur: >38°C oder <36°C
- Herzfrequenz: >90/min
- Atemfrequenz: >20/min oder paCO2 <32mmHg (<4,3kPa)
- Leukozyten: >12.000 Zellen/μl oder <4.000 Zellen/μl oder ≥10% unreife Neutrophile
Die Sepsis ist definiert durch eine systemische inflammatorische Reaktion, die nachweislich durch einen infektiösen Fokus verursacht wird.
Die schwere Sepsis geht einher mit Hypoperfusion, Hypotonie oder Organdysfunktion, was sich als Laktatazidose, Oligurie oder akute Änderung der Bewusstseinslage äußern kann. Als sepsisinduzierte Hypotonie wird ein systolischer Blutdruck von <90mmHg oder ein Abfall des systolischen Blutdruckes von ≥40mmHg vom Ausgangswert bezeichnet. Besteht die Hypotension mit Hypoperfusion und Organdysfunktion trotz adäquater Volumensubstitution weiter, so spricht man vom septischen Schock.
Ein weiterer oft benutzter Begriff ist das Multiorganversagen, was das gleichzeitige oder in rascher zeitlicher Abfolge auftretende Versagen von mindestens zwei Organsystemen beinhaltet.
Tab. 1: Sepsisdefinition entsprechend der Konsensuskonferenz 2001*
I. Nachweis einer Infektion oder Verdacht auf eine Infektion + einige der folgenden Kriterien:
Allgemeine Parameter
- Fieber (>38,3°C) oder Hypothermie (<36°C)
- Tachykardie (90/min oder >2-fache der Standardabweichung vom Normalwert
- Tachypnoe (>30/min)
- Eingeschränkter neurologischer Status
- Ödeme oder positive Flüssigkeitsbilanz (>20ml/kg/24h)
- Hyperglykämie (BZ >120mg/dl oder 7,7mM/l
Inflammatorische Parameter
- Leukozytose (>12.000/μl) oder Leukopenie (<4000/μl) oder Normwert mit >10% unreifen Formen
- C-reaktives Protein (>2fache Standardabweichung über Normwert)
- Prokalzitonin (>2fache Standardabweichnung über Normwert)
Hämodynamische Parameter
- Hypotonie (systol. Blutdruck <90mmHg, mittlerer arterieller Blutdruck <40mmHg oder Abfall <2fach Standardabweichung unter Normwert)
- Gemischtvenöse Sättigung <70%
- Herzindex <3,5l/min/m2
Organdysfunktion
- Arterielle Hypoxämie (paO2/FiO2 <300)
- Akute Oligurie (<0,5ml/kg/h oder 45mmol/l für ≥2h)
- Gerinnungsstörung (INR >1,5 oder PTT >60s)
- Hyperbilirubinämie (Bilirubin >4mg/dl oder >70mmol/l)
- Ileus (Fehlen von Darmgeräuschen)
- Kreatininanstieg um ≥0,5mg/dl
- Thrombozytopenie (<100.000/μl)
Gestörte Gewebeperfusion
- Erhöhtes Laktat (1mmol/l)
- Reduzierte kapilläre Füllung oder Marmorierung
*[2, 3]
Tab. 2: Sepsisdefinition und Schweregrad entsprechend der S-2-Leitlinie der Deutschen Sepsis-Gesellschaft und der Deutschen Interdisziplinären Vereinigung für Intensiv- und Notfallmedizin 2005*
I. Nachweis einer Infektion über den mikrobiologischen Nachweis oder durch klinische Kriterien
II. Systemic Inflammatory Response Syndrome (SIRS, mindestens 2 Kriterien)
- Fieber (>38,3°C) oder Hypothermie (<36°C)
- Tachykardie ≥90/min
- Tachypnoe (≥30/min) oder Hyperventilation (paCO2≤4,3kPa bzw. ≤33mmHg)
- Leukozytose (≥12.000/mm3) oder Leukopenie (≤4000/mm3) oder ≥10% unreife Neutrophile im Differenzialblutbild
III. Akute Organdysfunktion (mindestens 1 Kriterium)
- Akute Enzephalopathie: eingeschränkte Vigilanz, Desorientiertheit, Unruhe, Delirium
- Relative oder absolute Thrombozytopenie: Abfall der Thrombozyten >30% innerhalb von 24h oder Thrombozytenzahl ≤100.000mm3
- Arterielle Hypoxämie: paO2 ≤10kPa (≤75mmHg) unter Raumluft oder paO2(FiO2-Verhältnis ≤33kPa (≤250mmHg) unter Sauerstoffapplikation
- Renale Dysfunktion: Diurese ≤0,5ml/kg/h für wenigstens 2h trotz ausreichender Volumensubstitution und/oder Serumkreatininanstieg >2-fach oberhalb des üblichen Referenzbereichs
- Metabolische Azidose: Base Excess ≤-5mmol/l oder Laktatkonzentration >1,5-fach oberhalb des üblichen Referenzbereichs
Einteilung der Schweregrade:
Sepsis: Vorliegen der Kriterien I und II
Schwere Sepsis: Vorleigen der Kriterien I, II und III
Septischer Schock: Vorliegen der Kriterien I und II sowie für wenigstens 1h ein systolischer arterieller Blutdruck ≤90mmHg bzw. ein arterieller Mitteldruck ≤65mmHg oder notwendiger Vasopressoreneinsatz, um den systolischen arteriellen Blutdruck ≥90mmHg oder den arteriellen Mitteldruck ≥65mmHg zu halten. Die Hypotonie besteht trotz adäquater Volumengabe und ist nicht durch andere Ursachen zu erklären.
*[3, 12]
Sepsis – ein mehrphasiger Verlauf
Lange Zeit wurde die Sepsis mit ihren Folgeerscheinungen fast ausschließlich als eine über das normale physiologische Maß hinausschießende proinflammatorische Aktivierung des Immunsystems mit kaskadenartiger Amplifikation als eine Antwort auf einen infektiologischen bzw. gewebeschädigenden Stimulus angesehen. Hierbei kam es zu einer Dissoziation der physiologischen Antwort auf den pathogenen Stimulus mit dem Ergebnis der Entkoppelung der Abwehrreaktion von der eigentlichen Krankheitsursache [13, 14]. Die Antiinflammation als physiologischer Gegenspieler fand in dieser Betrachtungsweise keine oder nur eine marginale Beachtung.
Gegenwärtig ist man diesem Konzept jedoch abgerückt und geht aktuell von einem eher biphasischen Verlauf mit einer initialen proinflammatorischen Reaktion (SIRS) gefolgt von einer antiinflammatorischen Phase als Kompensation der SIRS (Compensatory anti-inflammatory response syndrome (CARS)) aus [15, 16]. Die pro- und anti-inflammatorischen Phasen können jedoch auch parallel verlaufen. In diesem Falle spricht man dann von einem Mixed antagonists response syndrome (MARS) [17, 18].
[/dt_sc_fullwidth_section]
Physiologie und Pathophysiologie des Immunsystems
Ein Entzündungsprozess wird durch einen lokal begrenzten Insult unterschiedlichster Genese ausgelöst. Das angeborene Immunsystem mit seinem Phagozytosesystem wird in dieser Kaskade durch sogenannte Danger associated molecular patterns (DAMPs) aktiviert. Unter DAMPs werden spezifische Strukturproteine von bakteriellen Pathogenen (pathogen associated molecular patterns (PAMPs), z.B. bakterielles Lipopolysaccharid) und Alarmine, die aus affektierten bzw. aus geschädigtem Gewebe freigesetzt werden, subsumiert. Die zellulären Komponenten des angeborenen Immunsystems (z.B. dendritische Zellen, Makrophagen/Monozyten, Granulozyten) erkennen die DAMPs über Pattern recognition receptors (PRR) (z.B. Toll-like-Rezeptor 4, TLR4) und lösen damit zum einen die Phagozytose dieser Pathogene aus und zum anderen eine intrazelluläre Signalkaskade mit dem Ergebnis einer gesteigerten Transkription von proinflammatorischen und regulatorischen Genen. Diese Genhochregulation führt wiederum zu einer vermehrten Ausschüttung von Zytokinen und Chemokinen [19-25].
Ziel der Phagozytose von pathogenen Strukturen mit anschließender Zelllyse und Prozessierung, sowie der Freisetzung von Zytokinen bzw. Chemokinen ist die Rekrutierung weiterer Abwehrzellen und die Unterbindung einer weiteren systemischen Ausbreitung.
Die professionellen Phagozyten können durch die Prozessierung der DAMPs wiederum als sogenannte antigenpräsentierende Zellen agieren und einen Brückenschlag zwischen dem angeborenen und adaptiven (erworbenen) Immunsystem bewirken [26]. Dabei werden Proteinfragmente der prozessierten DAMPs als Epitope über spezielle Moleküle (HLA-Komplex) den naiven T-Zellen des adaptiven Immunsystems präsentiert und durch kostimulatorische Moleküle eine adäquate Zell-Zellinteraktion hervorgerufen [27-29]. Aus den naiven T-Zellen entwickeln sich in Abhängigkeit der Epitop-Präsentation über das HLA-Molekül und der konsekutiven Zytokinausschüttung verschiedene T-Helfer-Subpopulationen [30, 31], die wiederum mit den ebenfalls zum erworbenen Immunsystems gehörenden B-Zellen eine „immunologische Synapse“ ausbilden [32]. Hierdurch kommt es zur B-Zellproliferation und Antikörpersynthese. Die Antikörper haben multiple Fähigkeiten, die von neutralisierend, opsonierend über agglutinierend bis hin zur Verbesserung der Phagozytose und Zytotoxizität reichen können [27, 33, 34].
Pathophysiologie der Sepsis
Die hier geschilderten Prozesse des menschlichen Immunsystems sind in ihrer Komplexität unabdingbar für das tägliche Überleben des menschlichen Organismus. Hierbei ist jedoch ein Gleichgewicht zwischen den pro- und antiinflammatorischen Gegenspielern essentiell, da ein Abweichen von der Homöostase zu der Entstehung pathologischer Zustände führt.
Beim SIRS bzw. bei der Sepsis wird dieses Gleichgewicht verloren und es kommt zu einer Dissoziation der physiologischen Antwort auf einen pathogenen Stimulus mit der Folge der vollständigen Entkoppelung der immunologischen Antwort von der eigentlichen Krankheitsursache. Die Relation zwischen Stimulus und Antwort ist dabei nicht mehr gegeben.
Ein lokal begrenzter Insult von unterschiedlichster Genese führt zur Aktivierung des angeborenen Immunsystems mit Brückenschlag zum erworbenen Immunsystem. Wird ein bestimmter Schweregrad des lokalen Insults überschritten, bleibt die physiologische inflammatorische Antwort nicht mehr lokalisiert, sondern es kommt zu einer systemischen Ausbreitung mit Aggravierung der immunologischen Antwort im Sinne einer systemischen Ausschüttung von proinflammatorischen Zytokinen (z.B. TNFα oder IL-6) und Chemokinen (z.B. MCP-1) [35, 36]. Nachfolgend kommt es zu einer Rekrutierung weiterer immunkompetenter Zellen, zur Aktivierung von Endothelzellen mit Hochregulierung von endothelialen und zellulären Adhäsionsmolekülen um die Diapedese der aktivierten Immunzellen zu verbessern, zur Aktivierung der Gerinnungskaskade und zu einer gesteigerten Produktion von Proteasen und Sauerstoffradikalen [23, 36-39]. Parallel dazu werden antiinflammatorische Zytokine (z.B. IL-10, IL-12) aber auch Akute-Phase-Proteine, Cortisol und Katecholamine freigesetzt [17, 18, 20, 38, 40-43].
Darüber hinaus kommt es im weiteren Verlauf zu einer Apoptose von dendritischen Zellen und Lymphozyten, wodurch eine immunologische Anergie und somit eine erhöhte Suszeptibilität für nosokomiale Infektionen entsteht [13, 44-50]. Im Gegensatz dazu ist die Apoptoserate von Makrophagen und neutrophilen Granulozyten herabgesetzt bei einer gleichzeitig überschießenden Produktion und Freisetzung von Proteasen und Sauerstoffradikalen. Die durch proinflammatorische Zytokine bedingte Aktivierung des Endothelsystems mit der vermehrten Expression von Adhäsionsmolekülen und einer ebenfalls gesteigerten Produktion von Stickstoffmonoxid (NO) führen zum einen zu einer Endotheldysfunktion sowie im weiteren Verlauf zu einer Apoptoseinduktion. Des weiteren wird die Homöostase des Gerinnungssystems zu Gunsten der prokoagulatorischen Faktoren verschoben, was im schlimmsten Fall zu einer Verbrauchskoagulopathie (disseminierte intravasale Gerinnungsstörung, DIC) führen kann. In diesem Fall liegen gleichzeitig eine vermehrte Thrombusbildung und eine erhöhte Blutungsneigung durch den Verbrauch von Gerinnungsfaktoren vor [38, 40, 51, 52]. Die Aktivierung des Endothelsystems und Gerinnungssystems führt schlussendlich zu einer endothelialen Permeabilitätsstörung mit Austritt von intravasaler Flüssigkeit in den Extrazellularraum und zur Ausbildung von Mikrothromben in der Gefäßendstrombahn. Dadurch kommt es zu einer konsekutiven systemischen Hypotension mit unzureichender Organperfusion und somit zu einer verminderten Sauerstoffversorgung. Aggraviert wird diese Organschädigung noch durch die mitochondriale Dysfunktion auf Grund der vermehrt gebildeten Sauerstoffradikale und Stickstoffmonoxidmoleküle [53, 54]. An diesem Punkt ist die Endstrecke der Sepsis mit Ausbildung eines Multiorganversagens (MODS) erreicht.
Klinisch äußert sich dieses Multiorganversagen durch ein pulmonale Globalinsuffizienz mit Hyperkapnie und Hypoxämie bis hin zum Acute Respiratory Distress Syndrome (ARDS) durch die gesteigerte Gefäßpermeabilität mit einhergehender Ödembildung[55]. Renal kommt es zum akuten anurischen Nierenversagen durch die Hypotension mit konsekutiver Hypoperfusion, vermehrter Zellapoptose und zytotoxischen Effekten [38, 40]. Das kardiovaskuläre System wird zum einen durch die Sticksoffmonoxid bedingte Hypotension mit Alteration der Vor- und Nachlast sowie durch eine allgemeine myokardiale Dysfunktion kompromittiert [40].
Besonderheiten der abdominellen Sepsis
Das Intestinum fand lange Zeit keine adäquate Berücksichtigung in der Pathogenese der Sepsis, bis mehrere wissenschaftliche Arbeiten aufzeigen konnten, dass der Darm durch eine mögliche „bakterielle Translokation“ nicht nur ein „stiller Teilhaber“ ist, sondern vielmehr einen „Motor“ in der Entstehung der Sepsis darstellen kann [56-58]. Die Translokation von Bakterien und Endotoxinen ist bei kritisch kranken Patienten durch eine epitheliale Schrankenstörung möglich und führt zu einer Sepsis mit Multiorganversagen [59-61]. Die genauen pathophysiologischen Mechanismen, die zu der epithelialen Barrierestörung führen, sind bis heute noch nicht hinreichend geklärt. Es ist bekannt, dass proinflammatorische Zytokine wie TNFα eine Desintegration von Junktionsproteinen zwischen den einzelnen Enterozyten bedingen [62]. Weitere Gründe für eine gestörte Barrierefunktion könnten aber auch Alterationen der physiologischen Darmflora sein, z.B. durch die Anwendung von Breitband-Antibiotika [63, 64].
Ein weiterer und wahrscheinlich viel bedeutenderer pathophysiologischer Mechanismus ist die im Rahmen der Sepsis auftretende relative Hypoperfusion durch einen erhöhten Sauerstoffbedarf und den intravasalen Flüssigkeitsverlust auf Grund der Endotheldysfunktion. Dadurch kommt es zu Epithelnekrosen und einem konsekutiven Schleimhautödem. In der Folge wird eine massive Zytokinproduktion generiert und es kommt zu einer Aktivierung von im Darm assoziierten lymphatischen Gewebe (GALT) beheimateten neutrophilen Granulozyten [65-68]. Über den Ductus thoracicus erreichen die freigesetzten Zytokine die systemische Makrozirkulation und in enger Nachbarschaft dazu auch die pulmonale Mikrozirkulation. Dadurch kann es trotz eines initial intraabdominellen Fokus zu einem Lungenversagen kommen [69-71].
Das Abdomen bietet jedoch nicht nur die hier geschilderten zellbiologischen Besonderheiten, sondern auch beachtenswerte anatomische Gegebenheiten. So wird die Bauchhöhle durch das parietale und viszerale Peritoneum ausgekleidet und schafft somit durch die Anheftungsstellen am Mesenterium und an den sekundär retroperitoneal gelegenen Organen miteinander kommunizierende Räume. Zusätzlich dient das Omentum majus als eine Erweiterung des angeborenen Immunsystems und ist in der Lage intraabdominelle Foci in den vom Peritoneum geschaffenen Räumen zu begrenzen und somit eine systemische Ausbreitung zu verhindern [72].
Einteilung der abdominellen Sepsis
Eine abdominelle Sepsis kann als eine intraabdominelle Sepsis mit extraperitonealer Begleitreaktion definiert werden, wobei sich die intraabdominelle Infektion klinisch als Peritonitis zeigt [72]. In Abhängigkeit der Pathogenese wird zwischen einer primären (nach hämato- oder lymphogener Streuung, z.B. spontan bakterielle Peritonitis), sekundären (bei Hohlorganperforation oder Abszessen, z.B. perforierte Sigmadivertikulitis) und tertiären Peritonitis (Persistenz der Infektion trotz Fokussanierung) unterschieden. Zudem kann die Peritonitis auch noch nach der Qualität des intraabdominellen Sekrets (z.B. gallig, fibrinös, putride, kotig) differenziert werden [73-75].
Eine weitere Einteilung der abdominellen Sepsis berücksichtigt den biphasischen Verlauf der Sepsis mit den daraus resultierenden immunologischen Besonderheiten der Immunparalyse durch ein Übergewicht von antiinflammatorischen Zytokinen, einer Apoptose von immunkompetenten Zellen und einer Reduktion der monozytären HLA-DR-Expression in der zweiten Phase [76-79]. Elektive chirurgische Eingriffe führen ebenfalls in ähnlicher Art und Weise durch das chirurgische Trauma zu einer Immunparalyse. Kommt es in dieser Phase zur Ausbildung einer Peritonitis, ist die Generierung einer adäquaten Immunantwort nicht mehr möglich und so eine erhöhte Letalität die Folge. Auf Grundlage dieser infektionsimmunologischen Besonderheiten wurde die abdominelle Sepsis bei sekundärer Peritonitis durch Maier et al. in eine Typ A- (spontaneously acquired abdominal sepsis, SAAS) und Typ B-Sepsis (postoperatively acquired abdominal sepsis, PAAS) klassifiziert [72]. Bei der Typ B-Sepsis liegt trotz eines zweiten infektiologischen Stimulus eine supprimierte Immunantwort vor auf Grund der vorausgegangenen Operation. Ein klassisches Beispiel für dieses Szenario ist die postoperative Anastomoseninsuffizienz.
Therapie der Sepsis
Die Grundpfeiler der Sepsistherapie basieren auf der Fokussanierung und der antimikrobiellen Therapie, wobei die letztgenannte Therapieoption durch die Progredienz der Antibiotikaresistenzen immer mehr an ihre Grenzen stößt. Alle weiteren Therapieansätze können nur noch als supportiv oder gar nur als adjunktiv und nicht mehr als kausal angesehen werden [12]. Der Einsatz kreislaufunterstützender Medikamente sowie die Volumensubstitution dienen als supportive Maßnahmen zur Aufrechterhaltung eines ausreichend hohen mittleren arteriellen Drucks und sollen somit das gefürchtete Multiorganversagen verhindern. Darüber hinaus spielen weitere Organersatzverfahren wie die differenzierte maschinelle Beatmung oder die Hämodialyse eine gewichtete Rolle.
Die Fokussanierung kann heutzutage nicht nur auf chirurgischem Wege erfolgen, sondern auch durch ein interventionelles Vorgehen (z.B. perkutane Drainagenanlage) gelöst werden.


Literatur
- Budelmann, G., [Hugo Schottmuller, 1867-1936. The problem of sepsis]. Internist (Berl), 1969. 10(3): p. 92-101.
- Levy, M.M., et al., 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med, 2003. 31(4): p. 1250-6.
- Moerer, O. and M. Quintel, [Sepsis in adult patients - definitions, epidemiology and economic aspects]. Internist (Berl), 2009. 50(7): p. 788, 790-4, 796-8.
- Beale, R., et al., Promoting Global Research Excellence in Severe Sepsis (PROGRESS): lessons from an international sepsis registry. Infection, 2009. 37(3): p. 222-32.
- Martin, G.S., et al., The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med, 2003. 348(16): p. 1546-54.
- Angus, D.C., et al., Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med, 2001. 29(7): p. 1303-10.
- Alberti, C., et al., Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med, 2003. 168(1): p. 77-84.
- Esper, A. and G.S. Martin, Is severe sepsis increasing in incidence AND severity? Crit Care Med, 2007. 35(5): p. 1414-5.
- Engel, C., et al., Epidemiology of sepsis in Germany: results from a national prospective multicenter study. Intensive Care Med, 2007. 33(4): p. 606-18.
- Bone, R.C., et al., Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest, 1992. 101(6): p. 1644-55.
- Bone, R.C., W.J. Sibbald, and C.L. Sprung, The ACCP-SCCM consensus conference on sepsis and organ failure. Chest, 1992. 101(6): p. 1481-3.
- Reinhart, K., et al., [Diagnosis and therapy of sepsis: guidelines of the German Sepsis Society Inc. and the German Interdisciplinary Society for Intensive and Emergency Medicine]. Anaesthesist, 2006. 55 Suppl 1: p. 43-56.
- Hauber, H.P. and P. Zabel, [Pathophysiology and pathogens of sepsis]. Internist (Berl), 2009. 50(7): p. 779-80, 782-4, 786-7.
- Hotchkiss, R.S. and I.E. Karl, The pathophysiology and treatment of sepsis. N Engl J Med, 2003. 348(2): p. 138-50.
- Oberholzer, A., C. Oberholzer, and L.L. Moldawer, Sepsis syndromes: understanding the role of innate and acquired immunity. Shock, 2001. 16(2): p. 83-96.
- Lederer, J.A., M.L. Rodrick, and J.A. Mannick, The effects of injury on the adaptive immune response. Shock, 1999. 11(3): p. 153-9.
- Adib-Conquy, M. and J.M. Cavaillon, Compensatory anti-inflammatory response syndrome. Thromb Haemost, 2009. 101(1): p. 36-47.
- Werdan, K., Pathophysiology of septic shock and multiple organ dysfunction syndrome and various therapeutic approaches with special emphasis on immunoglobulins. Ther Apher, 2001. 5(2): p. 115-22.
- Rittirsch, D., M.A. Flierl, and P.A. Ward, Harmful molecular mechanisms in sepsis. Nat Rev Immunol, 2008. 8(10): p. 776-87.
- Cohen, J., The immunopathogenesis of sepsis. Nature, 2002. 420(6917): p. 885-91.
- Bianchi, M.E., DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol, 2007. 81(1): p. 1-5.
- Cavaillon, J.M. and M. Adib-Conquy, Monocytes/macrophages and sepsis. Crit Care Med, 2005. 33(12 Suppl): p. S506-9.
- Brown, K.A., et al., Neutrophils in development of multiple organ failure in sepsis. Lancet, 2006. 368(9530): p. 157-69.
- Takeda, K., T. Kaisho, and S. Akira, Toll-like receptors. Annu Rev Immunol, 2003. 21: p. 335-76.
- Medzhitov, R., P. Preston-Hurlburt, and C.A. Janeway, Jr., A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature, 1997. 388(6640): p. 394-7.
- Satthaporn, S. and O. Eremin, Dendritic cells (I): Biological functions. J R Coll Surg Edinb, 2001. 46(1): p. 9-19.
- Schütt, C. and B. Bröker, Grundwissen Immunologie. Vol. 1. Auflage. 2006, München: Elsevier Spektrum Akademischer Verlag.
- Hoebe, K., E. Janssen, and B. Beutler, The interface between innate and adaptive immunity. Nat Immunol, 2004. 5(10): p. 971-4.
- Banchereau, J., et al., Immunobiology of dendritic cells. Annu Rev Immunol, 2000. 18: p. 767-811.
- Hsieh, C.S., et al., Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science, 1993. 260(5107): p. 547-9.
- Zhu, J. and W.E. Paul, CD4 T cells: fates, functions, and faults. Blood, 2008. 112(5): p. 1557-69.
- McHeyzer-Williams, L.J. and M.G. McHeyzer-Williams, Antigen-specific memory B cell development. Annu Rev Immunol, 2005. 23: p. 487-513.
- Tarlinton, D., B-cell memory: are subsets necessary? Nat Rev Immunol, 2006. 6(10): p. 785-90.
- LeBien, T.W. and T.F. Tedder, B lymphocytes: how they develop and function. Blood, 2008. 112(5): p. 1570-80.
- Boontham, P., et al., Surgical sepsis: dysregulation of immune function and therapeutic implications. Surgeon, 2003. 1(4): p. 187-206.
- Blackwell, T.S. and J.W. Christman, Sepsis and cytokines: current status. Br J Anaesth, 1996. 77(1): p. 110-7.
- Bone, R.C., Sepsis and the systemic inflammatory response syndrome (SIRS). Journal of Endotoxin Research, 1995. 2(3): p. 151-155.
- Paterson, R.L. and N.R. Webster, Sepsis and the systemic inflammatory response syndrome. J R Coll Surg Edinb, 2000. 45(3): p. 178-82.
- Marshall, J.C., Neutrophils in the pathogenesis of sepsis. Crit Care Med, 2005. 33(12 Suppl): p. S502-5.
- Abraham, E. and M. Singer, Mechanisms of sepsis-induced organ dysfunction. Crit Care Med, 2007. 35(10): p. 2408-16.
- Cavaillon, J.M., et al., Immunodepression in sepsis and SIRS assessed by ex vivo cytokine production is not a generalized phenomenon: a review. J Endotoxin Res, 2001. 7(2): p. 85-93.
- Munford, R.S. and J. Pugin, Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am J Respir Crit Care Med, 2001. 163(2): p. 316-21.
- Opal, S.M. and V.A. DePalo, Anti-inflammatory cytokines. Chest, 2000. 117(4): p. 1162-72.
- Green, D.R. and H.M. Beere, Apoptosis. Gone but not forgotten. Nature, 2000. 405(6782): p. 28-9.
- Hotchkiss, R.S., et al., Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med, 1999. 27(7): p. 1230-51.
- Hotchkiss, R.S., et al., Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A, 1999. 96(25): p. 14541-6.
- Hotchkiss, R.S., et al., Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol, 2002. 168(5): p. 2493-500.
- Hotchkiss, R.S., et al., Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol, 2001. 166(11): p. 6952-63.
- Venet, F., et al., Increased circulating regulatory T cells (CD4(+)CD25 (+)CD127 (-)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med, 2009. 35(4): p. 678-86.
- Venet, F., et al., Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25- lymphocytes. Crit Care Med, 2004. 32(11): p. 2329-31.
- Aird, W.C., The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood, 2003. 101(10): p. 3765-77.
- Aird, W.C., Endothelium as a therapeutic target in sepsis. Curr Drug Targets, 2007. 8(4): p. 501-7.
- Levi, M., E. de Jonge, and T. van der Poll, New treatment strategies for disseminated intravascular coagulation based on current understanding of the pathophysiology. Ann Med, 2004. 36(1): p. 41-9.
- Rudiger, A., M. Stotz, and M. Singer, Cellular processes in sepsis. Swiss Med Wkly, 2008. 138(43-44): p. 629-34.
- Kollef, M.H. and D.P. Schuster, The acute respiratory distress syndrome. N Engl J Med, 1995. 332(1): p. 27-37.
- Carrico, C.J., et al., Multiple-organ-failure syndrome. Arch Surg, 1986. 121(2): p. 196-208.
- Marshall, J.C., N.V. Christou, and J.L. Meakins, The gastrointestinal tract. The "undrained abscess" of multiple organ failure. Ann Surg, 1993. 218(2): p. 111-9.
- Wolochow, H., G.J. Hildebrand, and C. Lamanna, Translocation of microorganisms across the intestinal wall of the rat: effect of microbial size and concentration. J Infect Dis, 1966. 116(4): p. 523-8.
- Alexander, J.W., et al., The process of microbial translocation. Ann Surg, 1990. 212(4): p. 496-510; discussion 511-2.
- Deitch, E.A., Bacterial translocation of the gut flora. J Trauma, 1990. 30(12 Suppl): p. S184-9.
- Deitch, E.A., The role of intestinal barrier failure and bacterial translocation in the development of systemic infection and multiple organ failure. Arch Surg, 1990. 125(3): p. 403-4.
- Turner, J.R., Intestinal mucosal barrier function in health and disease. Nat Rev Immunol, 2009. 9(11): p. 799-809.
- van der Waaij, D., J.M. Berghuis-de Vries, and L.-v. Lekkerkerk, Colonization resistance of the digestive tract in conventional and antibiotic-treated mice. J Hyg (Lond), 1971. 69(3): p. 405-11.
- Banwell, J.G., et al., Intestinal microbial flora after feeding phytohemagglutinin lectins (Phaseolus vulgaris) to rats. Appl Environ Microbiol, 1985. 50(1): p. 68-80.
- Biffl, W.L. and E.E. Moore, Splanchnic ischaemia/reperfusion and multiple organ failure. Br J Anaesth, 1996. 77(1): p. 59-70.
- Deitch, E.A., Role of the gut lymphatic system in multiple organ failure. Curr Opin Crit Care, 2001. 7(2): p. 92-8.
- Rombeau, J.L. and J. Takala, Summary of round table conference: gut dysfunction in critical illness. Intensive Care Med, 1997. 23(4): p. 476-9.
- Swank, G.M. and E.A. Deitch, Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J Surg, 1996. 20(4): p. 411-7.
- Deitch, E.A., Gut-origin sepsis: evolution of a concept. Surgeon, 2012. 10(6): p. 350-6.
- Deitch, E.A., et al., Evidence favoring the role of the gut as a cytokine-generating organ in rats subjected to hemorrhagic shock. Shock, 1994. 1(2): p. 141-5.
- Magnotti, L.J., et al., Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg, 1998. 228(4): p. 518-27.
- Maier, S., et al., [Special aspects of abdominal sepsis]. Chirurg, 2005. 76(9): p. 829-36.
- Berger, D. and K. Buttenschoen, Management of abdominal sepsis. Langenbecks Arch Surg, 1998. 383(1): p. 35-43.
- Marshall, J.C. and M. Innes, Intensive care unit management of intra-abdominal infection. Crit Care Med, 2003. 31(8): p. 2228-37.
- Marshall, J.C., et al., Source control in the management of severe sepsis and septic shock: an evidence-based review. Crit Care Med, 2004. 32(11 Suppl): p. S513-26.
- Caille, V., et al., Histocompatibility leukocyte antigen-D related expression is specifically altered and predicts mortality in septic shock but not in other causes of shock. Shock, 2004. 22(6): p. 521-6.
- Tschoeke, S.K. and L.L. Moldawer, Human leukocyte antigen expression in sepsis: what have we learned? Crit Care Med, 2005. 33(1): p. 236-7.
- Ertel, W., et al., Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood, 1995. 85(5): p. 1341-7.
- Tschaikowsky, K., et al., Coincidence of pro- and anti-inflammatory responses in the early phase of severe sepsis: Longitudinal study of mononuclear histocompatibility leukocyte antigen-DR expression, procalcitonin, C-reactive protein, and changes in T-cell subsets in septic and postoperative patients. Crit Care Med, 2002. 30(5): p. 1015-23.