
 Wound Healing
Wound Healing Infection
Infection Acute Abdomen
Acute Abdomen Abdominal trauma
Abdominal trauma Ileus
Ileus Hernia
Hernia- General Surgery
 Benign Struma
Benign Struma Thyroid Carcinoma
Thyroid Carcinoma Hyperparathyroidism
Hyperparathyroidism Hyperthyreosis
Hyperthyreosis Adrenal Gland Tumors
Adrenal Gland Tumors- Endocrine Surgery
 Achalasia
Achalasia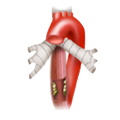 Esophageal Carcinoma
Esophageal Carcinoma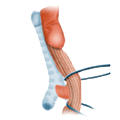 Esophageal Diverticulum
Esophageal Diverticulum Esophageal Perforation
Esophageal Perforation Corrosive Esophagitis
Corrosive Esophagitis Gastric Carcinoma
Gastric Carcinoma Peptic Ulcer Disease
Peptic Ulcer Disease GERD
GERD Bariatric Surgery
Bariatric Surgery- Upper GI-Tract
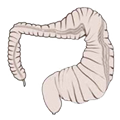 CIBD
CIBD Divertikulitis
Divertikulitis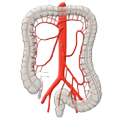 Colon Carcinoma
Colon Carcinoma Proktology
Proktology Rectal Carcinoma
Rectal Carcinoma- Lower GI-Tract
 Anatomy
Anatomy Ikterus
Ikterus Cholezystolithiais
Cholezystolithiais Benign Liver Lesions
Benign Liver Lesions Malignant Liver Leasions
Malignant Liver Leasions Pancreatitis
Pancreatitis Pancreatic carcinoma
Pancreatic carcinoma- Hepatobiliary Surgery
Infection, Sepsis, Intensive Therapy
Infection is defined as invasion of the body by microorganisms inducing the simultaneous localized or generalized defensive reaction of the body.
The invading pathogens can be divided into obligate pathogens and facultative pathogens. Obligate pathogens always cause disease symptoms, and do not belong to the normal body flora; Salmonella typhi is an example of an obligate pathogen. Facultative pathogens belong to the normal bowel flora and do not cause disease symptoms; Escherichia coli are a type of facultative pathogen. Only if they leave this milieu and enter a wound, for example, do they cause an infection.
A different classification distinguishes between endogenous and exogenous as well as opportunistic pathogens. An example of the latter is Pseudomonas aeruginosa, which opportunistically exploits weakened immune systems; in healthy immune systems, no infection occurs.

Morphology
Morphologically, infections can be classified according to their appearance. Some infections have nonspecific morphologies, like phlegmon, or Staphylococcus aureus-induced abscesses. An abscess is defined as a localized purulent destruction of tissue separated by a pyogenic membrane from surrounding organ structures. A phlegmon is a deep tissue infection that spreads diffusely and is thus poorly delimited. Empyema, i.e. the accumulation of pus in an existing body cavity, is an infection with nonspecific morphology.
Among infections with specific morphology are granulomas in Mycobacterium tuberculosis, cutaneous gummas in Treponema pallidum, and abscesses (Drusen) in actinomycete infections. There are also pathogens that form special endotoxins which can trigger specific reactions in the body. These include the Clostridia, whose subgroup C. tetani causes tetanus, C. perfringens, which causes gas gangrene, and C. diphtheria.

Opisthotonus in a patient suffering from tetanus – Painting by Charles Bell 1809
‘Lockjaw’ is a disease that has all but disappeared from consciousness in our latitudes, since it is rarely seen due to the high rate of immunization. In developing nations, however, many people die of tetanus infections. In surgical treatment of the wound, the medical history and, if appropriate, updating of the tetanus-immunization status are important components. Tetanus is triggered by Clostridium tetani, an obligate anaerobe bacterium that is extremely resistant in its spore form and ubiquitous in soil. The bacterial form is able to form various exotoxins, including tetanospasmin and tetanolysin. The bacteria travel from the wound via axonal transport mechanisms to the anterior horns of the spinal cord, where they trigger the typical symptoms of tetanus: trismus (lockjaw), risus sardonicus (spasm of the facial muscles), spontaneous muscle spasms, opisthotonus (arching of the head, neck, and back). A manifest tetanus infection can lead via paralysis of the pharyngeal muscles to asphyxia and death. Untreated the disease is usually fatal; a survived infection confers no immunity. Therapeutically, extensive excision of the wound and open wound care are important. High-dose antibiotics are given, a mixed infection is often present. In addition, immediately active immunization with tetanus adsorbate (Tetanol) and administration of tetanus-hyperimmunoglobulin (Tetagam) should be done. Depending on the severity of symptoms, sedation or muscle relaxation with ventilation therapy may be needed.
– Hugo Schottmüller
“Sepsis is present if a focus has developed from which pathogenic bacteria, constantly or periodically, invade the blood stream in such a way that this causes subjective and objective symptoms.”
Sepsis
The concept of sepsis was mentioned already in the age of Hippocrates, but was only given a precise definition by Hugo Schottmüller in 1914: “Sepsis is present if a focus has developed from which pathogenic bacteria, constantly or periodically, invade the blood stream in such a way that this causes subjective and objective symptoms.” [1]
Gastbeitrag
 This topic was posted by Dr. Flemming.
This topic was posted by Dr. Flemming.
Sepsis – One of the Most Common Causes of Death
Sepsis is a feared complication in clinical routine and is still — despite continuous advances in medical research, diagnosis, and therapy — associated with very high morbidity and mortality. In non-cardiological units in Germany, sepsis is the most common cause of death [2-4]. Due to the epidemiological transition in the population, increase in the number of indications for invasive procedures, and the progression of antimicrobial-resistance and immunosuppressive procedures, the incidence of sepsis has grown year by year [5, 6]. The Competence Network Sepsis (SepNet) calculates that sepsis has a prevalence of 12.4%, severe sepsis and septic shock of 11%. Depending on the study and severity, the mortality varies between 20% and 60% [5-9]. Respiratory tract infection is the most common cause of a severe septic clinical picture, followed by intra-abdominal infections (focus). Other causes of severe sepsis are catheter and/or foreign body-associated infections, surgical infections, invasive candidiasis in immunocompromised patients, and bacterial meningitis.
Systemic Inflammatory Response Syndrome (SIRS)? Sepsis? – Definitions of a clinical picture
Following the initial definition of sepsis by Hugo Schottmüller, sepsis is now defined in light of current knowledge as follows: „Sepsis is caused by pathogenic microorganisms and their products and leads to endogenous synthesis and secretion of humoral and cellular mediators. Unlike in infection, in sepsis the host is typically not successful in limiting the inflammatory reaction locally.“ [3]
At the Consensus Conference of the American College of Chest Physicians and the Society of Critical Care Medicine in 1991, a more extensive classification and definition of sepsis was worked out. Over time it was recognized that not only infectious stimuli, but also traumata, burns, intoxications, and ischemia can give rise to a septic clinical picture [10, 11]. A distinction is made between a systemic inflammatory reaction (systemic inflammatory response syndrome, SIRS), sepsis, severe sepsis, and septic shock. A SIRS is characterized by the presence of at least two of the four following criteria:
- Body temperature: >38°C oder <36°C
- Heart rate: >90/min
- Respiratory rate: >20/min or paCO2 <32mmHg (<4,3kPa)
- Leukocytes: >12.000 cells/μl or <4.000 cells/μl or ≥10% immature neutrophils
Sepsis is defined by a systemic inflammatory reaction, which is caused by a focus of infection.
Severe sepsis is accompanied by hypoperfusion, hypotension, organ dysfunction, which can be expressed as lactic acidosis (←Laktatazidose -?), oliguria, and acute changes in mental status. Hypotension with a systolic blood pressure of <or a drop in the systolic blood pressure of ≥40mmHg from baseline values is considered to be sepsis-induced hypotension. If hypotension with hypoperfusion and organ dysfunction persists despite adequate volume substitution, the patient has septic shock.
Another common term is multiple organ failure, which designates the simultaneous or rapidly sequential failure of at least two organ systems.
Table 1: Sepsis Definition According to the Consensus Conference 2001*
I. I. Demonstration of an infection or suspicion of an infection + several of the following criteria:
General parameters
- Fever (>38,3°C) or hypothermia (<36°C)
- Tachycardia (90/min or >2-fold the standard deviation from the normal value
- Tachypnea (>30/min)
- Impaired neurological status
- Edema or positive fluid balance (>20ml/kg/24h)
- Hyperglycemia (BS >120mg/dl or 7,7mM/l
Inflammatory Parameters
- Leukocytosis (>12.000/μl) or leukopenia (<4000/μl) or normal value with >10% immature forms
- C-reactive protein (>2-fold standard deviation above normal value)
- Procalcitonin (>2-fold standard deviation above normal value)
Hemodynamic Parameters
- Hypotension (systolic blood pressure <90mmHg, mean arterial blood pressure <40mmHg or decrease <2-fold standard deviation above normal value)
- Mixed venous [oxygen] saturation <70%
- Heart index <3,5l/min/m2
Organ dysfunction
- Arterial hypoxemia (paO2/FiO2 <300)
- Acute oliguria (<0,5ml/kg/h or 45mmol/l für ≥2h)
- Coagulation disorder (INR >1,5 or PTT >60s)
- Hyperbilirubinemia (Bilirubin >4mg/dl or >70mmol/l)
- Ileus (no bowel sounds)
- Creatinine increase of about ≥0,5mg/dl
- Thrombocytopenia (<100.000/μl)
Impaired Tissue Perfusion
- Hyperlactatemia (1mmol/l)
- Decreased capillary refill or mottling *[2, 3]
Table 2: Sepsis definition and severity according to the S-2 Guidelines of the German Sepsis Society and the German Interdisciplinary Association of Intensive Care and Emergency Medicine 2005*
I. Confirmation of infection – Diagnosis of an infection on the basis of microbiological evidence or clinical criteria
II. Systemic inflammatory host response (SIRS) (at least 2 criteria)
- Fever (>38,3°C) or hypothermia (<36°C) confirmed by rectal, intravascular or intravesical measurement
- Tachycardia: heart rate ≥90/min
- Tachypnea {frequency (≥20/min) or hyperventilation (paCO2≤4,3kPa bzw. ≤33mmHg)
- Leukocytosis (≥12.000/mm3) or leukopenia (≤4000/mm3) or ≥10% uimmature neutrophils in differential blood count
III. Acute organ dysfunction (at least 1 criterion)
- Acute encephalopathy: reduced alertness, disorientation, agitation, delirium
- Relative or absolute thrombocytopenia: decrease in platelet counts by more than 30% within 24 hours or a platelet count of less than lOO.OOD/mm^. Thrombocytopenia due to acute hemorrhage or immunological causes must be ruled out.
- Arterial hypoxemia: paO2 ≤10kPa (≤75mmHg) while breathing ambient air or a PaOj/FiO; ratio ≤33kPa (≤250mmHg) on oxygen administration. A clinically manifest heart or lung disease must be ruled out as a cause of hypoxemia.
- Renal impairment: diuresis of ≤0,5ml/kg/h for at least 2 hours despite adequate volume resuscitation and/or an increase in serum creatinine level to 3-fold twice the upper limit of normal (ULN)
- Metabolic acidosis: Base excess of ≤-5mmol/l o or lactate concentration of >1,5x ULN
Grading:
Sepsis: criteria I and II
Severe sepsis: criteria I, II and III
Septic shock: criteria I and II, as well as a systolic arterial blood pressure of £90 mmHg for at least 1 hour, or mean arterial pressure of £65 mmHg, or the necessity of vasopressor administration to maintain a target systolic arterial pressure of >90 mmHg or mean arterial pressure of >65mmHg. Hypotension persists despite adequate volume resuscitation and cannot be explained by other causes.
*[3, 12]
Sepsis – A Multiphase Course
Sepsis with its sequelae was long regarded as a proinflammatory activation of the immune system that overshoots the normal physiological range (Maß) with a cascade-like amplification in response to an infectious and/or tissue-damaging stimulus. The dissociation of the physiological response from the pathogenic stimulus results in a uncoupling of the defense reaction from the actual cause of disease [13, 14]. An anti-inflammatory phase as a physiological counterresponse (Gegenspieler) found in this view no or only marginal consideration.
Today, however, this concept has been abandoned in favor of a biphasic course, with an initial proinflammatory reaction (SIRS) followed by an anti-inflammatory phase as compensation for the SIRS (compensatory anti-inflammatory response syndrome [CARS]) [15, 16]. The pro- and anti-inflammatory phases can also run parallel to one another, in which case the term mixed antagonist response syndrome (MARS) is applied [17, 18].
[/dt_sc_fullwidth_section]
Physiology and Pathophysiology of the Immune System
An inflammatory process is triggered by locally confined insults of diverse etiology. In this cascade, the innate immune system with its mononuclear phagocytic system is activated by so-called danger-associated molecular patterns (DAMPs). The term DAMPs encompasses specific structural proteins of bacterial pathogens (pathogen-associated molecular patterns [PAMPs], e.g. bacterial lipopolysaccharide) and alarmins, which are released from the affected or damaged tissue. The cellular component of the innate immune system (e.g. dendritic cells, macrophages/monocytes, granulocytes) recognizes DAMPs by pattern recognition receptors (PRR) (e.g. toll-like receptor 4, TLR4) and trigger the phagocytosis of these pathogens and an intracellular signal cascade that results in enhanced transcription of proinflammatory and regulatory genes. This gene upregulation leads in turn to increased secretion of cytokines and chemokines [19-25].
The goal of phagocytosis of pathogenic structures with subsequent cell lysis, processing, and release of cytokines and chemokines is the recruitment of additional defense cells and prevention of further systemic spread.
Professional phagocytes can act by processing DAMPs as so-called antigen-presenting cells and thus bridge the gap between the innate and acquired immune systems [26]. Protein fragments of the processed DAMPs are presented as epitopes via (über) special molecules (HLA complex) to the naive T cells of the acquired (adaptive) immune system and an adequate cell-cell interaction is induced by costimulatory molecules [27-29]. Various T-helper subpopulations develop from naive T cells in dependence on epitope presentation via HLA molecules and subsequent cytokine secretion [30, 31], which in turn forms an „immunological synapse“ with B cells, which are also part of the acquired immune system [32]. The result is B-cell proliferation and antibody synthesis. The antibodies thus produced have multiple capabilities ranging from neutralization, opsonization, to agglutination and improvement of phagocytosis and cytotoxicity [27, 33, 34].
Pathophysiology of Sepsis
The functions of the highly complex immune system are indispensable to the survival of the human organism. A balance between the pro- and anti-inflammatory responses is essential, since a deviation from homeostasis can lead to pathological conditions.
In SIRS and sepsis this balance is lost and the physiological response becomes dissociated from the pathogenic stimulus, with a consequent complete uncoupling of the immunologic response from the actual cause of disease. The relation between stimulus and response in lost.
If the local insult exceeds a certain severity, the physiological inflammatory response does not remain localized, but the immunological response spreads systemically through a systemic release of proinflammatory cytokines (e.g. TNFα or IL-6) and chemokines (e.g. MCP-1) [35, 36]. Subsequently, additional immunocompetent cells are recruited, endothelial cells are activated with upregulation of endothelial and cell-adhesion molecules that improve diapedesis of the activated immune cells, the coagulation cascade is activated, and protease and oxygen-radical production increases [23, 36-39]. Parallel to this, anti-inflammatory cytokines (e.g. IL-10, IL-12), but also acute-phase proteins, cortisol, and catecholamine, are released [17, 18, 20, 38, 40-43].
In further course, apoptosis of dendritic cells and lymphocytes occurs, giving rise to an immunological anergia and thus to an elevated susceptibility to nosocomial infections [13, 44-50]. Counter to this, the apoptosis rate of macrophages and
neutrophilic granulocytes is lowered with a simultaneous excess production and release of proteases and oxygen radicals. Proinflammatory cytokines activate the endothelial system with enhanced expression of adhesion molecules and elevated production of nitrogen monoxide (NO), leading to endothelial dysfunction and in further course to apoptosis. Furthermore, homeostasis of the coagulation system is shifted in favor of procoagulatory factors, which in the worst case can lead to disseminated intravascular coagulation (DIC). This involves the simultaneous presence of increased thrombus formation and an elevated bleeding tendency secondary to clotting factors being consumed [38, 40, 51, 52]. Activation of the endothelial and coagulation systems leads ultimately to an endothelial permeability disorder with leakage of intravascular fluids into the extracellular space and formation of microthrombi in the vascular microcirculation. The result is consecutive systemic hypotension with inadequate organ perfusion and consequent diminished oxygen supply. This organ damage is further aggravated by the mitochondrial dysfunction due to increased formation of oxygen radicals and nitrogen monoxide molecules [53, 54]. At this point the end phase of sepsis begins with onset of multiorgan dysfunction syndrome (MODS).
Clinically, MODS is expressed as a pulmonary global insufficiency with hypercapnia, hypoxemia, and acute respiratory distress syndrome (ARDS) due to (durch) the increased vascular permeability with accompanying edema [55]. Acute anuric kidney failure occurs due to the hypotension with consequent hypoperfusion, increased cell apoptosis, and cytotoxic effects [38, 40]. The cardiovascular system is compromised both by nitrogen oxide-induced hypotension with alteration of the pre- and afterload and by general myocardial dysfunction [40].
Characteristics of Abdominal Sepsis
The intestine was long a neglected factor in the pathogenesis of sepsis. Then several scientific studies showed that the intestine by means of a possible „bacterial translocation“ may be not just a „silent partner“, but even a „motor“ driving the development of sepsis [56-58]. The translocation of bacteria and endotoxins is possible in critically ill patients due to an epithelial blood-aqueous-barrier disorder and leads to sepsis with MODS [59-61]. The exact pathophysiological mechanisms that lead to epithelial barrier disorder are still not adequately understood. Proinflammatory cytokines such as TNFα are known to cause a disintegration of junction proteins between the individual enterocytes [62]. The disturbed barrier function could also be caused by alterations in the physiological bowel flora due e.g. to the use of broad-spectrum antibiotics [63, 64].
A further and probably much more important pathophysiological mechanism is the sepsis-associated relative hypoperfusion due to an increased oxygen demand and intravascular fluid loss secondary to endothelial dysfunction. This leads to epithelial necrosis and consequent mucous membrane edema. There is a subsequent massive production of cytokines and activation of neutrophilic granulocytes residing in the gut-associated lymphatic tissue (GALT) [65-68]. The released cytokines reach the systemic macrocirculation and pulmonary microcirculation via the thoracic duct leading, despite the initial intra-abdominal focus, to respiratory failure [69-71].
The abdomen, however, offers more than just the described cell biology features, but also some noteworthy anatomic conditions. The abdominal cavity, for example, is lined by the parietal and visceral peritoneum, creating interconnecting spaces at the attachment points (Anheftungsstellen) on the mesenterium and secondarily retroperitoneal organs. In addition, the greater omentum serves as an extension of the innate immune system and can restrict intra-abdominal foci to the peritoneal spaces and thus hinder systemic spread [72].
Classification of Abdominal Sepsis
An abdominal sepsis can be defined as an intra-abdominal sepsis with accompanying extraperitoneal reaction, the intra-abdominal infection presenting clinically as peritonitis [72]. Depending on the pathogenesis, a distinction is made between a primary peritonitis (hematogenous or lymphogenous spread, e.g. spontaneous bacterial peritonitis), secondary peritonitis (hollow organ perforation or abscesses, e.g. perforated sigma diverticulitis) and tertiary peritonitis (persistence of infection despite eradication of the infectious focus. In addition, the peritonitis can be differentiated according to the nature of the intra-abdominal secretion (e.g. bilious, fibrinous, putrid, feculent) [73-75].
A further classification of abdominal sepsis considers its biphasic course and the resulting immunological characteristics of immune paralysis caused by the preponderance of anti-inflammatory cytokines, apoptosis of immunocompetent cells in the first phase, and the reduction in monocytic HLA-DR expression in the second [76-79]. The surgical trauma of elective surgery can lead in a similar manner to immune paralysis. If a peritonitis develops in this phase, an adequate immune response is no longer possible with consequent increased lethality. Based on these immunological characteristics of infection, abdominal sepsis in secondary peritonitis is divided by Maier et al. into a type A sepsis (spontaneously acquired abdominal sepsis, SAAS) and a type B sepsis (postoperatively acquired abdominal sepsis, PAAS) [72]. In type B sepsis, despite a second infectious stimulus there is a muted immune response due to the preceding surgery. A classical example for this scenario is postoperative anastomotic insufficiency.
Treatment of Sepsis
The cornerstones of sepsis therapy are eradication of the septic focus and antimicrobial treatment; the latter is increasingly losing its effectiveness due to the growth of antimicrobial resistance. All other treatment approaches can be regarded as supportive or merely adjunctive and can no longer be regarded as curative [12]. Cardiovascular drugs or volume substitution are supportive measures to maintain an adequate mean arterial pressure and thus to prevent the dreaded multiple organ failure. Other organ replacement methods such as differentiated mechanical respiration and hemodialysis also play an important role.
Today septic foci can be eradicated (Fokussanierung) not only by surgical means but also by interventional procedures (e.g. percutaneous drainage).


References
- Budelmann, G., [Hugo Schottmuller, 1867-1936. The problem of sepsis]. Internist (Berl), 1969. 10(3): p. 92-101.
- Levy, M.M., et al., 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med, 2003. 31(4): p. 1250-6.
- Moerer, O. and M. Quintel, [Sepsis in adult patients – definitions, epidemiology and economic aspects]. Internist (Berl), 2009. 50(7): p. 788, 790-4, 796-8.
- Beale, R., et al., Promoting Global Research Excellence in Severe Sepsis (PROGRESS): lessons from an international sepsis registry. Infection, 2009. 37(3): p. 222-32.
- Martin, G.S., et al., The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med, 2003. 348(16): p. 1546-54.
- Angus, D.C., et al., Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med, 2001. 29(7): p. 1303-10.
- Alberti, C., et al., Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med, 2003. 168(1): p. 77-84.
- Esper, A. and G.S. Martin, Is severe sepsis increasing in incidence AND severity? Crit Care Med, 2007. 35(5): p. 1414-5.
- Engel, C., et al., Epidemiology of sepsis in Germany: results from a national prospective multicenter study. Intensive Care Med, 2007. 33(4): p. 606-18.
- Bone, R.C., et al., Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest, 1992. 101(6): p. 1644-55.
- Bone, R.C., W.J. Sibbald, and C.L. Sprung, The ACCP-SCCM consensus conference on sepsis and organ failure. Chest, 1992. 101(6): p. 1481-3.
- Reinhart, K., et al., [Diagnosis and therapy of sepsis: guidelines of the German Sepsis Society Inc. and the German Interdisciplinary Society for Intensive and Emergency Medicine]. Anaesthesist, 2006. 55 Suppl 1: p. 43-56.
- Hauber, H.P. and P. Zabel, [Pathophysiology and pathogens of sepsis]. Internist (Berl), 2009. 50(7): p. 779-80, 782-4, 786-7.
- Hotchkiss, R.S. and I.E. Karl, The pathophysiology and treatment of sepsis. N Engl J Med, 2003. 348(2): p. 138-50.
- Oberholzer, A., C. Oberholzer, and L.L. Moldawer, Sepsis syndromes: understanding the role of innate and acquired immunity. Shock, 2001. 16(2): p. 83-96.
- Lederer, J.A., M.L. Rodrick, and J.A. Mannick, The effects of injury on the adaptive immune response. Shock, 1999. 11(3): p. 153-9.
- Adib-Conquy, M. and J.M. Cavaillon, Compensatory anti-inflammatory response syndrome. Thromb Haemost, 2009. 101(1): p. 36-47.
- Werdan, K., Pathophysiology of septic shock and multiple organ dysfunction syndrome and various therapeutic approaches with special emphasis on immunoglobulins. Ther Apher, 2001. 5(2): p. 115-22.
- Rittirsch, D., M.A. Flierl, and P.A. Ward, Harmful molecular mechanisms in sepsis. Nat Rev Immunol, 2008. 8(10): p. 776-87.
- Cohen, J., The immunopathogenesis of sepsis. Nature, 2002. 420(6917): p. 885-91.
- Bianchi, M.E., DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol, 2007. 81(1): p. 1-5.
- Cavaillon, J.M. and M. Adib-Conquy, Monocytes/macrophages and sepsis. Crit Care Med, 2005. 33(12 Suppl): p. S506-9.
- Brown, K.A., et al., Neutrophils in development of multiple organ failure in sepsis. Lancet, 2006. 368(9530): p. 157-69.
- Takeda, K., T. Kaisho, and S. Akira, Toll-like receptors. Annu Rev Immunol, 2003. 21: p. 335-76.
- Medzhitov, R., P. Preston-Hurlburt, and C.A. Janeway, Jr., A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature, 1997. 388(6640): p. 394-7.
- Satthaporn, S. and O. Eremin, Dendritic cells (I): Biological functions. J R Coll Surg Edinb, 2001. 46(1): p. 9-19.
- Schütt, C. and B. Bröker, Grundwissen Immunologie. Vol. 1. Auflage. 2006, München: Elsevier Spektrum Akademischer Verlag.
- Hoebe, K., E. Janssen, and B. Beutler, The interface between innate and adaptive immunity. Nat Immunol, 2004. 5(10): p. 971-4.
- Banchereau, J., et al., Immunobiology of dendritic cells. Annu Rev Immunol, 2000. 18: p. 767-811.
- Hsieh, C.S., et al., Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science, 1993. 260(5107): p. 547-9.
- Zhu, J. and W.E. Paul, CD4 T cells: fates, functions, and faults. Blood, 2008. 112(5): p. 1557-69.
- McHeyzer-Williams, L.J. and M.G. McHeyzer-Williams, Antigen-specific memory B cell development. Annu Rev Immunol, 2005. 23: p. 487-513.
- Tarlinton, D., B-cell memory: are subsets necessary? Nat Rev Immunol, 2006. 6(10): p. 785-90.
- LeBien, T.W. and T.F. Tedder, B lymphocytes: how they develop and function. Blood, 2008. 112(5): p. 1570-80.
- Boontham, P., et al., Surgical sepsis: dysregulation of immune function and therapeutic implications. Surgeon, 2003. 1(4): p. 187-206.
- Blackwell, T.S. and J.W. Christman, Sepsis and cytokines: current status. Br J Anaesth, 1996. 77(1): p. 110-7.
- Bone, R.C., Sepsis and the systemic inflammatory response syndrome (SIRS). Journal of Endotoxin Research, 1995. 2(3): p. 151-155.
- Paterson, R.L. and N.R. Webster, Sepsis and the systemic inflammatory response syndrome. J R Coll Surg Edinb, 2000. 45(3): p. 178-82.
- Marshall, J.C., Neutrophils in the pathogenesis of sepsis. Crit Care Med, 2005. 33(12 Suppl): p. S502-5.
- Abraham, E. and M. Singer, Mechanisms of sepsis-induced organ dysfunction. Crit Care Med, 2007. 35(10): p. 2408-16.
- Cavaillon, J.M., et al., Immunodepression in sepsis and SIRS assessed by ex vivo cytokine production is not a generalized phenomenon: a review. J Endotoxin Res, 2001. 7(2): p. 85-93.
- Munford, R.S. and J. Pugin, Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am J Respir Crit Care Med, 2001. 163(2): p. 316-21.
- Opal, S.M. and V.A. DePalo, Anti-inflammatory cytokines. Chest, 2000. 117(4): p. 1162-72.
- Green, D.R. and H.M. Beere, Apoptosis. Gone but not forgotten. Nature, 2000. 405(6782): p. 28-9.
- Hotchkiss, R.S., et al., Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med, 1999. 27(7): p. 1230-51.
- Hotchkiss, R.S., et al., Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A, 1999. 96(25): p. 14541-6.
- Hotchkiss, R.S., et al., Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol, 2002. 168(5): p. 2493-500.
- Hotchkiss, R.S., et al., Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol, 2001. 166(11): p. 6952-63.
- Venet, F., et al., Increased circulating regulatory T cells (CD4(+)CD25 (+)CD127 (-)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med, 2009. 35(4): p. 678-86.
- Venet, F., et al., Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25- lymphocytes. Crit Care Med, 2004. 32(11): p. 2329-31.
- Aird, W.C., The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood, 2003. 101(10): p. 3765-77.
- Aird, W.C., Endothelium as a therapeutic target in sepsis. Curr Drug Targets, 2007. 8(4): p. 501-7.
- Levi, M., E. de Jonge, and T. van der Poll, New treatment strategies for disseminated intravascular coagulation based on current understanding of the pathophysiology. Ann Med, 2004. 36(1): p. 41-9.
- Rudiger, A., M. Stotz, and M. Singer, Cellular processes in sepsis. Swiss Med Wkly, 2008. 138(43-44): p. 629-34.
- Kollef, M.H. and D.P. Schuster, The acute respiratory distress syndrome. N Engl J Med, 1995. 332(1): p. 27-37.
- Carrico, C.J., et al., Multiple-organ-failure syndrome. Arch Surg, 1986. 121(2): p. 196-208.
- Marshall, J.C., N.V. Christou, and J.L. Meakins, The gastrointestinal tract. The “undrained abscess” of multiple organ failure. Ann Surg, 1993. 218(2): p. 111-9.
- Wolochow, H., G.J. Hildebrand, and C. Lamanna, Translocation of microorganisms across the intestinal wall of the rat: effect of microbial size and concentration. J Infect Dis, 1966. 116(4): p. 523-8.
- Alexander, J.W., et al., The process of microbial translocation. Ann Surg, 1990. 212(4): p. 496-510; discussion 511-2.
- Deitch, E.A., Bacterial translocation of the gut flora. J Trauma, 1990. 30(12 Suppl): p. S184-9.
- Deitch, E.A., The role of intestinal barrier failure and bacterial translocation in the development of systemic infection and multiple organ failure. Arch Surg, 1990. 125(3): p. 403-4.
- Turner, J.R., Intestinal mucosal barrier function in health and disease. Nat Rev Immunol, 2009. 9(11): p. 799-809.
- van der Waaij, D., J.M. Berghuis-de Vries, and L.-v. Lekkerkerk, Colonization resistance of the digestive tract in conventional and antibiotic-treated mice. J Hyg (Lond), 1971. 69(3): p. 405-11.
- Banwell, J.G., et al., Intestinal microbial flora after feeding phytohemagglutinin lectins (Phaseolus vulgaris) to rats. Appl Environ Microbiol, 1985. 50(1): p. 68-80.
- Biffl, W.L. and E.E. Moore, Splanchnic ischaemia/reperfusion and multiple organ failure. Br J Anaesth, 1996. 77(1): p. 59-70.
- Deitch, E.A., Role of the gut lymphatic system in multiple organ failure. Curr Opin Crit Care, 2001. 7(2): p. 92-8.
- Rombeau, J.L. and J. Takala, Summary of round table conference: gut dysfunction in critical illness. Intensive Care Med, 1997. 23(4): p. 476-9.
- Swank, G.M. and E.A. Deitch, Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J Surg, 1996. 20(4): p. 411-7.
- Deitch, E.A., Gut-origin sepsis: evolution of a concept. Surgeon, 2012. 10(6): p. 350-6.
- Deitch, E.A., et al., Evidence favoring the role of the gut as a cytokine-generating organ in rats subjected to hemorrhagic shock. Shock, 1994. 1(2): p. 141-5.
- Magnotti, L.J., et al., Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg, 1998. 228(4): p. 518-27.
- Maier, S., et al., [Special aspects of abdominal sepsis]. Chirurg, 2005. 76(9): p. 829-36.
- Berger, D. and K. Buttenschoen, Management of abdominal sepsis. Langenbecks Arch Surg, 1998. 383(1): p. 35-43.
- Marshall, J.C. and M. Innes, Intensive care unit management of intra-abdominal infection. Crit Care Med, 2003. 31(8): p. 2228-37.
- Marshall, J.C., et al., Source control in the management of severe sepsis and septic shock: an evidence-based review. Crit Care Med, 2004. 32(11 Suppl): p. S513-26.
- Caille, V., et al., Histocompatibility leukocyte antigen-D related expression is specifically altered and predicts mortality in septic shock but not in other causes of shock. Shock, 2004. 22(6): p. 521-6.
- Tschoeke, S.K. and L.L. Moldawer, Human leukocyte antigen expression in sepsis: what have we learned? Crit Care Med, 2005. 33(1): p. 236-7.
- Ertel, W., et al., Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood, 1995. 85(5): p. 1341-7.
- Tschaikowsky, K., et al., Coincidence of pro- and anti-inflammatory responses in the early phase of severe sepsis: Longitudinal study of mononuclear histocompatibility leukocyte antigen-DR expression, procalcitonin, C-reactive protein, and changes in T-cell subsets in septic and postoperative patients. Crit Care Med, 2002. 30(5): p. 1015-23.